Bieżący numer
Artykuły zaakceptowane
O czasopiśmie
Rada Naukowa
Kolegium Redakcyjne
Polityka prawno-archiwizacyjna
Kodeks etyki publikacyjnej
Wydawca
Informacja o przetwarzaniu danych osobowych w ramach plików cookies oraz subskrypcji newslettera
Archiwum
Dla autorów
Dla recenzentów
Kontakt
Recenzenci
Recenzenci rocznika 2025
Recenzenci rocznika 2024
Recenzenci rocznika 2023
Recenzenci rocznika 2022
Recenzenci rocznika 2021
Recenzenci rocznika 2020
Recenzenci rocznika 2019
Recenzenci rocznika 2018
Recenzenci rocznika 2017
Recenzenci rocznika 2016
Recenzenci rocznika 2015
Recenzenci rocznika 2014
Recenzenci rocznika 2013
Recenzenci rocznika 2012
Polecamy
Śląski Uniwersytet Medyczny w Katowicach
Sklep Wydawnictw SUM
Biblioteka Główna SUM
Polityka prywatności
Deklaracja dostępności
Recenzenci
Recenzenci rocznika 2025
Recenzenci rocznika 2024
Recenzenci rocznika 2023
Recenzenci rocznika 2022
Recenzenci rocznika 2021
Recenzenci rocznika 2020
Recenzenci rocznika 2019
Recenzenci rocznika 2018
Recenzenci rocznika 2017
Recenzenci rocznika 2016
Recenzenci rocznika 2015
Recenzenci rocznika 2014
Recenzenci rocznika 2013
Recenzenci rocznika 2012

Diagnostic problems of rare diseases — case report of amyloidosis
1
Department of Internal Medicine, Autoimmune Diseases and Diabetology, Faculty of Medical Sciences in Katowice, Medical University of Silesia, Katowice, Poland
Autor do korespondencji
Agnieszka Żak-Gołąb
ul. Medyków 14, 40-752 Katowice, Uniwersyteckie Centrum Kliniczne im. prof. K. Gibińskiego ŚUM
ul. Medyków 14, 40-752 Katowice, Uniwersyteckie Centrum Kliniczne im. prof. K. Gibińskiego ŚUM

SŁOWA KLUCZOWE
DZIEDZINY
STRESZCZENIE
Amyloidosis is a group of disorders characterised by the accumulation of insoluble
proteins in tissues. These deposits lead to organ dysfunction and, in many cases, death. This
paper discusses the case of a 61-year-old male patient who presented with fatigue, dyspnoea
with minimal exertion and noncharacteristic abdominal cramping pain that had been present for
three months. Laboratory tests showed abnormalities indicating cholestasis, liver and kidney
damage, and hypercalcemia. Echocardiography revealed thickening of the left ventricular walls
with preserved ejection function, a strongly hyperechoic interventricular septum. Despite
intensive pharmacotherapy, the patient developed multiorgan failure and died due to the signs
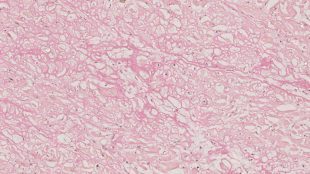
of hepatic encephalopathy and acute kidney injury. According to the autopsy report storage
disease is suspected, and additional examinations revealed intercellular amyloid deposits
(Congo Red +, Sinus Red +) in the heart muscle, spleen, and liver.
REFERENCJE (21)
1.
Cook J, Muchtar E, Warsame R. Updates in the Diagnosis and Management of AL Amyloidosis. Curr Hematol Malig Rep. 2020;15(3):155–167. doi: 10.1007/s11899-020-00574-5.
2.
Kumar N, Zhang NJ, Cherepanov D, Romanus D, Hughes M, Faller DV. Global epidemiology of amyloid light-chain amyloidosis. Orphanet J Rare Dis. 2022;17(1):278. doi: 10.1186/s13023-022-02414-6.
3.
Vaxman I, Gertz M. When to Suspect a Diagnosis of Amyloidosis. Acta Haematol. 2020;143(4):304–311. doi: 10.1159/000506617.
4.
McCausland KL, White MK, Guthrie SD, Quock T, Finkel M, Lousada I, et al. Light Chain (AL) Amyloidosis: The Journey to Diagnosis. Patient. 2018;11(2):207–216. doi: 10.1007/s40271-017-0273-5.
5.
Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30(9):989–995. doi: 10.1200/JCO.2011.38.5724.
6.
Kozak S, Ulbrich K, Migacz M, Szydło K, Mizia-Stec K, Holecki M. Cardiac Amyloidosis-Challenging Diagnosis and Unclear Clinical Picture. Medicina (Kaunas). 2021;57(5):450. doi: 10.3390/medicina57050450.
7.
Laires PA, Fang S, Evans J, Thompson J, Gaur A, Staruk B, et al. Incidence and prevalence of light chain amyloidosis in the United States in 2019–2021 using Optum EHR data. Sci Rep. 2025;15(1):25149. doi: 10.1038/s41598-025-09498-7.
8.
Grzeszczak W, Franek E, Szypowska A, Filipow W, Zięba M, Kabicz P, et al. Incidence of non-hereditary amyloidosis in Poland. Ann Acad Med Siles. 2021;75:99–106. doi: 10.18794/aams/141603.
9.
Sabinot A, Ghetti G, Pradelli L, Bellucci S, Lausi A, Palladini G. State-of-the-art review on AL amyloidosis in Western Countries: Epidemiology, health economics, risk assessment and therapeutic management of a rare disease. Blood Rev. 2023;59:101040. doi: 10.1016/j.blre.2023.101040.
10.
D’Souza A, Singh A, Szabo A, Lian Q, Pezzin L, Sparapani R. Timing and co-occurrence of symptoms prior to a diagnosis of light chain (AL) amyloidosis. Res Sq [Preprint]. 2024;rs.3.rs-3788661. doi: 10.21203/rs.3.rs-3788661/v1.
11.
Brandt K, Cathcart ES, Cohen AS. A clinical analysis of the course and prognosis of forty-two patients with amyloidosis. Am J Med. 1968;44(6):955–969. doi: 10.1016/0002-9343(68)90095-8.
12.
Faa G, Van Eyken P, De Vos R, Fevery J, Van Damme B, De Groote J, et al. Light chain deposition disease of the liver associated with AL-type amyloidosis and severe cholestasis. J Hepatol. 1991;12(1):75–82. doi: 10.1016/0168-8278(91)90913-v.
13.
Lee JG, Wilson JA, Gottfried MR. Gastrointestinal manifestations of amyloidosis. South Med J. 1994;87(2):243–247. doi: 10.1097/00007611-199402000-00019.
14.
Park MA, Mueller PS, Kyle RA, Larson DR, Plevak MF, Gertz MA. Primary (AL) hepatic amyloidosis: clinical features and natural history in 98 patients. Medicine. 2003;82(5):291–298. doi: 10.1097/01.md.0000091183.93122.c7.
15.
Garcia-Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, Brucato A, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021;42(16):1554–1568. doi: 10.1093/eurheartj/ehab072.
16.
Dittrich T, Kimmich C, Hegenbart U, Schönland SO. Prognosis and Staging of AL Amyloidosis. Acta Haematol. 2020;143(4):388–400. doi: 10.1159/000508287.
17.
Ebert EC, Nagar M. Gastrointestinal manifestations of amyloidosis. Am J Gastroenterol. 2008;103(3):776–787. doi: 10.1111/j.1572-0241.2007.01669.x.
18.
Shinohara M, Hashimoto M, Kitamura Y, Nakashima K, Hamaoka M, Miguchi M, et al. Preoperative diagnosis and safe surgical approach in gallbladder amyloidosis: a case report. Surg Case Rep. 2024;10(1):89. doi: 10.1186/s40792-024-01897-8.
19.
Cichoń M, Mizia-Stec K, Wojnicz R, Kukla P, Drożdż M. Cardiac amyloidosis: myocardial biopsy as a tool in chemotherapy implementation and sudden cardiac death prevention. Pol Arch Intern Med. 2018;128(4):254–255. doi: 10.20452/pamw.4244.
20.
Wang YD, Zhao CY, Yin HZ. Primary hepatic amyloidosis: a mini literature review and five cases report. Ann Hepatol. 2012;11(5):721–727. doi: 10.1016/S1665-2681(19)31450-4.
21.
Bucurica S, Nancoff AS, Moraru MV, Bucurica A, Socol C, Balaban DV, et al. Digestive Amyloidosis Trends: Clinical, Pathological, and Imaging Characteristics. Biomedicines. 2024;12(11):2630. doi: 10.3390/biomedicines12112630.
Udostępnij
ARTYKUŁ POWIĄZANY
| eISSN: | 1734-025X |

Śląski Uniwersytet Medyczny w Katowicach, jako Operator Serwisu annales.sum.edu.pl, przetwarza dane osobowe zbierane podczas odwiedzania Serwisu. Realizacja funkcji pozyskiwania informacji o Użytkownikach i ich zachowaniu odbywa się poprzez dobrowolnie wprowadzone w formularzach informacje, zapisywanie w urządzeniach końcowych plików cookies (tzw. ciasteczka), a także poprzez gromadzenie logów serwera www, będącego w posiadaniu Operatora Serwisu. Dane, w tym pliki cookies, wykorzystywane są w celu realizacji usług zgodnie z Polityką prywatności.
Możesz wyrazić zgodę na przetwarzanie danych w tych celach, odmówić zgody lub uzyskać dostęp do bardziej szczegółowych informacji.
Możesz wyrazić zgodę na przetwarzanie danych w tych celach, odmówić zgody lub uzyskać dostęp do bardziej szczegółowych informacji.